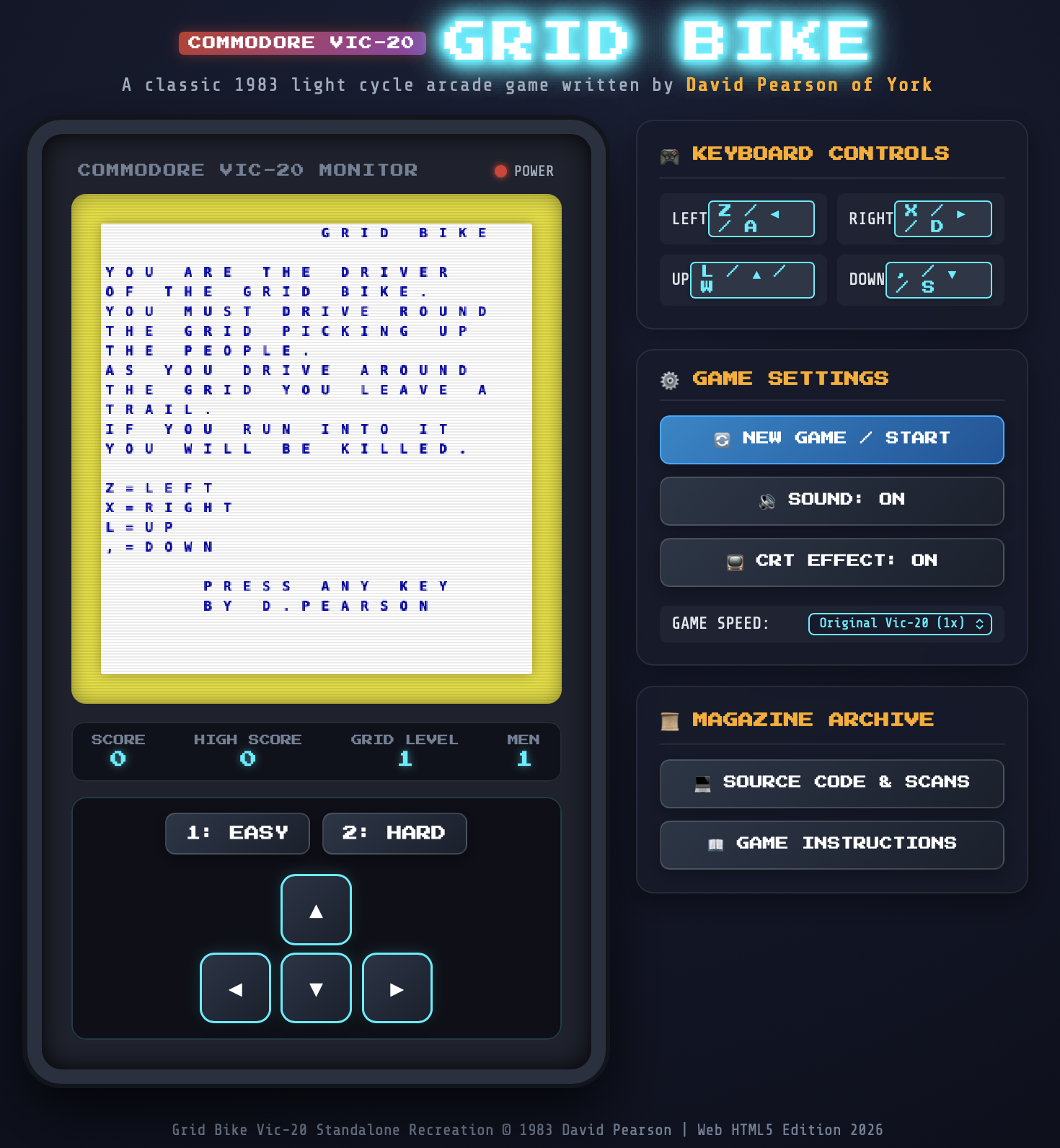
As Rogallo got close to being "stable", I did a fair bit of work on its documentation. Something that I wanted to include in the site was a good collection of up-to-date screenshots. These are all created on the fly. To do this, of course, requires something that looks like a Gemini server.
Initially, this was simple: Rogallo only supported Gemini capsules, so I could illustrate most things just using Gemtext files in the local filesystem (with an admonition in the documentation to point out that Rogallo was for more than viewing local files). This, of course, wasn't going to scale when I added Finger support, and neither was it going to work well for Gopher support.
The solution seemed obvious: use a lightweight local server for these protocols. With this need in place SmolServe was born. As mentioned when I released Rogallo v1.5.0, this isn't a project to build a comprehensive smolweb server. The aim is to build myself a minimal just-good-enough server that helps me with local testing and documentation.
As it stands, SmolServe supports Gemini, Gopher, Finger and Spartan. Or, rather, it supports a just-enough-to-get-by version of each of those protocols. None are implemented in a way that would serve as a "production" server; they're implemented to allow me to generate screenshots for the Rogallo documentation, involving any of the supported protocols, without the need to rely on services I don't control and which aren't local.
The big benefit of SmolServe is the exec support. With this, you can run up the server and then have it run another command. Once that command finishes its work, SmolServe will close down too. This means that, when it comes to producing the Rogallo documentation, I can just have the server running when I need it. Pulling some snippets from the Rogallo Makefile:
run := uv run
smol := $(run) smolserve --config $(docs)server/smolserve.toml
smolexec := $(smol) exec --
mkdocs := $(smolexec) mkdocs
##############################################################################
# Documentation.
.PHONY: docs
docs:
$(mkdocs) build
.PHONY: rtfm
rtfm:
$(mkdocs) serve --livereload
.PHONY: publishdocs
publishdocs: clean-docs
$(mkdocs) gh-deploy
The idea is that, when I build the documentation, I actually run smolserve, which in turn runs mkdocs, which then produces the documentation while the local server is available.
I also use this sort of approach for local testing of the content of my capsule that lives over on tilde.team.
.PHONY: view
view:
uv run smolserve --config smolserve.toml exec rogallo open gemini://localhost/
My aim now is that, if I add any other protocols to Rogallo, I'll add a just-good-enough version of them to SmolServe to help me with testing and documentation. For the moment, though, I think it's in a stable and usable state.